Design, Synthesis and Evaluation of small molecules
Focus
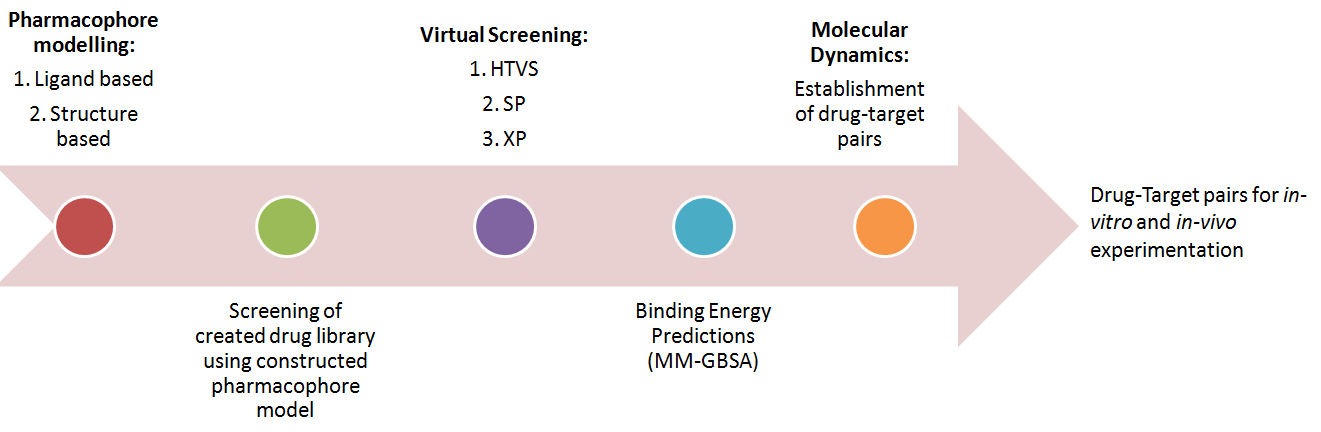
Advanced technology has paved way to implement computational strategies at several stages of drug discovery right from hit to lead identification, lead optimization and formulation design. During the process of drug design and discovery, it is crucial to understand the 3D structure, molecular mechanism, biochemical functions of target proteins. Plenty of crystallographic structures of target proteins are deposited in Protein Data Bank (PDB), millions of chemical compounds of both synthetic and natural origin are available in public databases and few databases share valuable information on outcomes of experimental studies. Particularly, natural compounds have been attracting huge attention because of their broad range of therapeutic properties with specificity in their action in human health care as functional foods, pharmaceuticals and nutraceuticals. Screening those chemical databases by different approaches like molecular docking, pharmacophore modelling, Induced-fit docking, Prime MM- GBSA calculations, Molecular Dynamics and ADMET predictions will aid us to identify in silico potential hit compounds and further validation through experimental in vitro or in vivo studies help us to identify safe and potential lead molecules for respective target proteins. Computational software’s are stepping towards development of in-house databases by adapting techniques like Reaction-based Enumeration and improving the accuracy of predictions with Artificial Intelligence and Machine Learning tools.
Working Hypothesis
Research Topics
- Cancer
- HIV
- Tuberculosis
Facilities Available
- Molecular docking
- Pharmacophore modelling
- 2D & 3D QSAR Model generation
- Homology modelling
- Chemical Synthesis



